
Comprehensive annotation and functional exploration of microRNAs in lettuce
Lettuce (Lactuca sativa), of the Asteraceae family, is a leafy vegetable with important economic value. According to the FAO (Food and Agriculture Organization, http://www.fao.org/), the global production of lettuce, together with chicory, exceeded 29 million tons in 2019, ranking third of all leafy vegetables. The worldwide popularity of lettuce is due to its diverse cultivars, taste, and high nutritional value, being rich in vitamins, folates, and minerals. With the availability of a reference genome and numerous transcriptomes, lettuce has become a model for studying Asteraceae and related plants. Lettuce has numerous cultivars, including butterhead, crisphead, looseleaf, romaine, stem, and many mixed types, which are highly varied in morphology, and provide good material for studying the morphological development of plants. The same is true for lettuce in terms of leaf colors, metabolites, heat responses, viral resistance, etc. Lettuce is also an important example of crop domestication and for adaptation research. Recently, lettuce has been used as a novel platform for biopharmaceutical production due to its numerous advantages, including low processing cost, consistent and scalable production, and the excellent biosafety profile of transgenic plants.
In recently, by sequencing sRNAs in four different tissues and using a pipeline centered by miRDeep-P2, 157 high-confidence miRNAs were identified in lettuce. The detailed characteristics of these miRNAs were annotated, including sequences, structure, conservation, clusters, selection after duplication, and expression patterns in different tissues. Furthermore, a regulatory network involving miRNAs, TFs, and targets, was constructed, and the connection between different feed forward loops (FFLs) helped in defining a number of new regulatory links.
This work systematically identified and annotated miRNAs in lettuce, and conducted preliminary explorations of the functions of these miRNAs by means of target genes, expression correlation, regulatory networks, etc., and lays a solid foundation for further research on lettuce miRNAs.
Comprehensive identification and annotation of miRNAs in lettuce
To comprehensively identify miRNAs in lettuce, sRNA libraries from different tissues, including leaf, stem, root and flower, were prepared in duplicate and observed with well consistency. These eight libraries produced 240 million raw reads and 235 million clean reads. Length distribution of these sRNA libraries showed two peaks at 21 and 24 nt in both clean and unique sRNAs, potentially reflecting the abundance of miRNAs and small interference RNAs (siRNAs), respectively (Figure 1AB). The large decrease at 21 nt from clean to unique read distributions further supports this speculation (Figure 1AB) because, in general, miRNAs prefer to accumulate many identical reads, while siRNAs possess much greater sequence diversity. Compared to other tissues, abundance at the 21-nt peak is higher than that at 24-nt in the flower (Figure 1A), suggesting higher miRNA activities in flower tissue.
MiRDeep-P2 was employed to identify miRNAs when only 19–25 nt reads were selected as inputs. Filtered with strict plant miRNA criteria, 157 miRNA loci, belonging to 67 families, were annotated. Several features of the miRNA loci were scanned, and most of their precursors, accounting for 80%, were less than 150 nts (Figure 1C), and the length of mature miRNAs were predominantly 21 nts (Figure 1D), while the first nucleic acid of mature miRNAs is frequently U (Figure 1E). All of these observations meet plant miRNA characteristics, as indicated by previous research, suggesting that the identification and annotation is reliable. A conservation search against all miRNA entries in PmiREN1.0, found that 109, including 27 families, are conserved since their counterparts could be found in other non-Asteraceae species, while only six are considered Asteraceae-specific, because counterparts were only detected in Asteraceae species. Forty-two items were annotated as lettuce specific, considering no conserved ones were detected (Figure 1F). Among the conserved families, miR395 has the most abundant members, while the other 18 families have multiple members (Figure 1G). The annotation for each miRNA includes all relevant information, including genomic location, sequences, secondary structure, and reads signature along precursors. Figures 1H-M, show conserved, Asteraceae-specific and lettuce-specific examples, respectively. Standard stem-loop structure and reads signature, such as the accumulation at mature and star miRNAs, strongly support them being highly confident candidates.
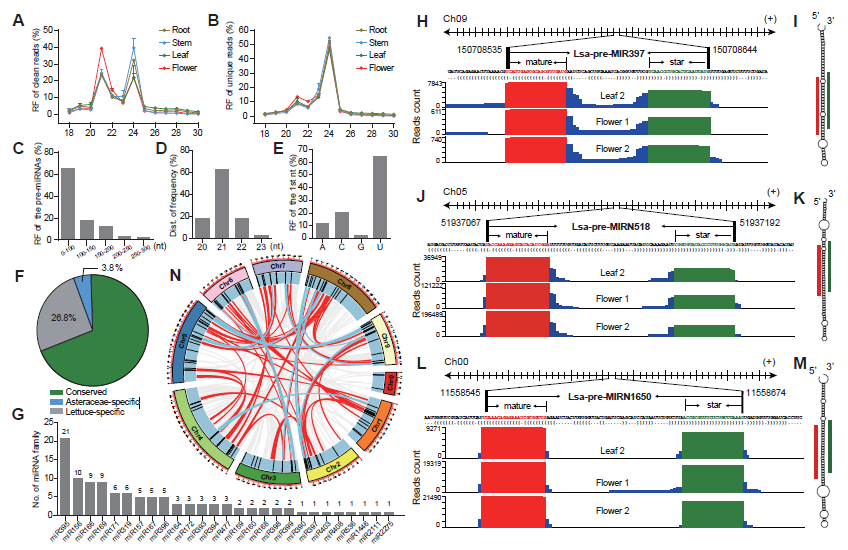
Figure 1. Identification and annotation of miRNAs in lettuce.
Regulatory network exploration of lettuce miRNAs
The regulatory elements of miRNAs and target genes do not exist independently, and further constitute a regulatory network. Taking into account the particularity of TFs, that is, they are important miRNA targets that can, in return, regulate miRNA expression, a regulatory network was explored, with TFs, miRNAs, and targets as basic elements. First, miRNA targets from 23 TF families were collected, then the promoter sequences of miRNAs and target genes were extracted, and by a search of known binding motifs of these 23 TFs, a preliminary regulatory relationship of TFs on miRNA and miRNA target genes was constructed. At the same time, using the predicted regulatory relationship between miRNAs and targets, the regulatory relationship between miRNA and target genes was determined (Figure 2A). In detail, 1,051 interactions were found between TFs and miRNAs, named TMIs, and 73,133 interactions identified between TFs and miRNA targets, so called TTIs. Together with the 6,540 interactions predicted between miRNAs and targets (MTIs), 46,083 cascades binding TMIs and MTIs were achieved, and 43,554 FFL motifs were constructed (Figure 2A). Based on these FFLs and the links crossing them, a complex regulatory network with TFs, miRNAs, and targets was constructed (Figure 2B).
The regulatory network was constructed to further understand the regulation between miRNAs, as the core, and a combination of TFs and target genes. In this network, any element centered on a specific miRNA can independently become a module. For example, miR408 accepts regulations from four TFs, and a dozen targets could be regulated by both miR408 and these four TFs (Figure 2C). As described previously, Lsa-miRN518 is an Asteraceae-specific miRNA, and its 30 targets can also be regulated by several TFs (Figure 2D). Figure 2E shows a lettuce-specific miRNA, Lsa-miRN46, which has a relatively simple FFL module. When the function of each gene was added into these modules, their functions could be further determined. Using Figure 2F as an example where all non-TFs and non-miRNA nodes were filtered out, the regulatory network uncovered numerous new links between miRNAs and TFs. In this two particles of the large network, regulations between MYB to miR156, miR156 to SBP, and miR396 to GRF and WRKY, and miR166 to HD-ZIP, have been well established by previous studies. However, this data supports that more TFs could regulate miR156, and the targets of miR156 could further regulate the modules of miR399 and miRN565. Moreover, more modules are involved in the regulation network of miR396 and miR166, forming a seven-layer network system. Based on the known functions of these miRNAs and TFs, these two network particles, could potentially be involved in development, circadian clock, flowering, response to insect damage and environmental challenges, etc.
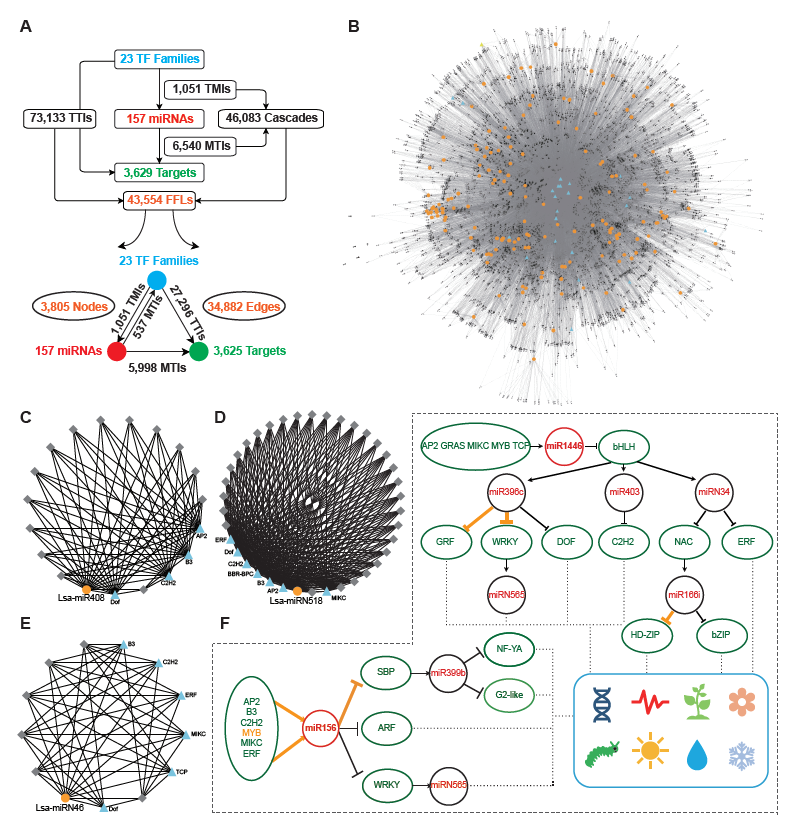
Figure 2. Construction of miRNA regulatory network in lettuce